During the stillness in the summer months I've revived an
old project regarding the construction of a new basis set optimized for calculating spin-spin coupling constants. It is the aug-cc-pVTZ-J basis set family I hope to contribute to and I am deriving basis sets for the elements
Gallium,
Germanium,
Arsenic,
Selenium and
Bromine.
I've already constructed the uncontracted version of the basis set (aptly named aug-cc-pVTZ-Juc) by saturating it with additional primitive s, p and d functions following the even-tempered approach were the ratio between exponents are kept constant. It is a pretty straightforward and standard way of increasing the size of a basis set.
If you do this for the simplest hydride of Bromine, I.e. HBr, you end up with something that looks like the following figure when calculating spin-spin coupling constants as the basis is saturated with tight s-functions (black), five tight s-functions and tight p-functions (medium grey) and five tight s-functions, two tight p-functions and tight d-functions (light gray):
It can be seen that there is a nice convergence as the basis set is saturated with s-functions (owing to a convergence of the Fermi contact term). Adding tight p- and d-functions are observed to yield a small increase and a bit of wobbling back and forth as convergence is reached.
I've settled on the the following primitive set of basis functions for the basis sets across all tested elements: [26s16p14d2f] which means I've added 5s, 2p and 4d primitive basis functions to the original uncontracted aug-cc-pVTZ basis set.
However, this is much too large to be useful in any sort of real calculation except for the very basic hydrides I'm using. So we need to contract the basis set. So far, the approach with the J-family of the aug-cc-pVTZ basis sets has been to use the molecular obital coefficients from the simple hydrides as contraction coefficients up to some level (I should rather use down to I suppose but it feels wrong).
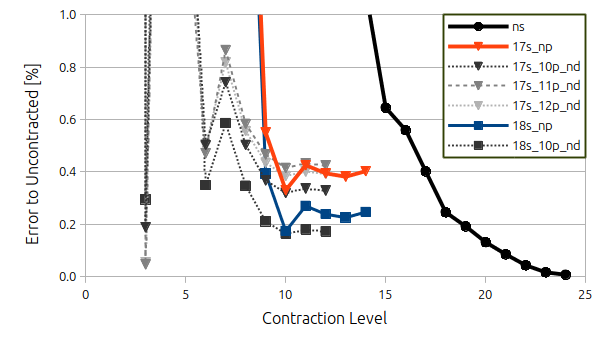
Contracting the 26 primitive s-functions first into ns contracted s-functions (while letting the p and d-functions remain uncontracted) gives the solid black line with cirles in the following figure where the error compared to the fully uncontracted basis set is given in percent.
Here we observe that we must use a rather loose form of contraction of at least 15 (I.e. the 26 primitives are fixed in such a way that there is only 15 contracted basis functions left) to obtain an error below 1 % compared to the uncontracted level.
I've furthermore shown data for using a contraction of either 17 or 18 for the
s-functions (to get a really low error) and contracted the
p-functions on top of that (orange for 17 and blue for 18, respectively). Here we observe that saturation in both cases is obtained by contracting the 16 primitive
p-functions into 10 contracted
p-functions. Notice that the difference between the orange and blue curve is equal to the difference between the error we make in the contraction of the
s-functions. Going above contraction level 10 for the p-functions is not giving us much improvement overall.
Finally, the dashed gray lines represent the contraction of
d-functions on top of either the 17
s10
p or 18
s10
p results. We observe that a contraction level of 10 is needed for convergence (although it could be argued that a contraction level 9 is more than enough). Again, the difference between 17
s10
p10
d and 18
s10
p10
d is equal to the error from the contraction of the
s-functions and
p-functions.
As a final note, we see from the 17
s11
p and 17
s12
p that using either of these as a base for the contraction of the
d-functions, the errors amounts to the difference betwen the contraction level of the
p-functions.