In the last blog
post, we were discussing both the saturation and the contraction. In
the end, I've settled on a (26s16p12d2f) basis set for the
aug-cc-pVTZ-Juc basis set (uc is for uncontracted) and now I turn to
the contraction of the basis set.
So why do we
contract the basis set? - well – for every primitive s-function
have in our system, we increase the size of the Fock matrix by one.
For p-functions, we have \(p_x\), \(p_y\) and \(p_z\) and thus for every primitive
p-function we increase the size of the Fock matrix by three. For
d-fucntions it's five and for f-functions is 10. So, by just
including one atom using our newly constructed aug-cc-pVTZ-Juc basis
set increases the size of the Fock matix by
\[ 26 \times 1 + 3 \times 16 + 12 \times 5 + 2 \times 10 = 156 \]
and remember that
the methods you want to use scales really bad with the number of
basis functions.
So a contraction of
the basis set can be seen as a reasonable reduction of the basis set
size without compromising its quality (too much).

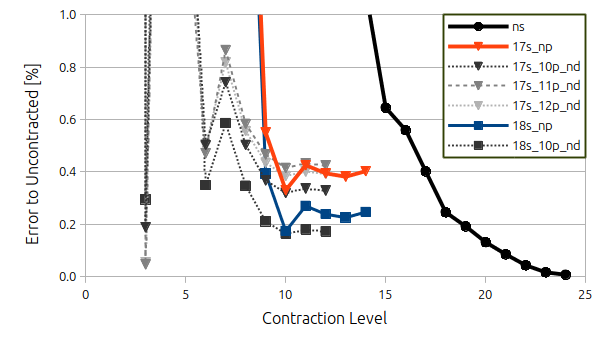
Above
you see a figure I 've made with the error in percent to
aug-cc-pVTZ-Juc as the contraction level is increased. The further
you go to the right, the more s-functions (see the number on the
x-axis) you have in your contracted basis set. Obviously, the fewer
the better because of computational speed but as you go further to
the left, the error starts increasing. Initially not so much, but
below 10 some errors are > 100 %.
I've decided that an
error below 1 % in the contraction when compared to the uncontracted
basis set is acceptable (at least for the s-functions), but as
observed from the figure above, this threshold is reached at quite
different points depending on the which element is investigated.
And this is where it
stops being: there is one answer only and it turns into voodoo. So
personally, I think that a contraction level of 14 would be quite
nice, but since that is the first point where all five elements are
below or very close to 1 % (Bromine is not quite there) I don't feel
that this should be the contraction level. Going to a contraction
level of 15, all calculations give results that are below 1 % but
somehow it saddens me that the error for Gallium shoots up to 1 %
while Bromine goes down. Obviously I should not concern myself with
Germanium, Arsenic or Selenium as their errors are well below 0.2 %
for the same contraction level. I guess for
s-functions I could probably settle with a contraction level of 17
since reduces the error to 0.6 % in the worst case (Gallium) without adding a lot of computational expense, but
then again – a difference of 0.4 % is not really a whole lot.